Publications
Publications in peer reviewed journals
Abundant toxin-related genes in the genomes of beneficial symbionts from deep-sea hydrothermal vent mussels.
2015 - Elife, e07966
Abstract:
Bathymodiolus mussels live in symbiosis with intracellular sulfur-oxidizing (SOX) bacteria that provide them with nutrition. We sequenced the SOX symbiont genomes from two Bathymodiolus species. Comparison of these symbiont genomes with those of their closest relatives revealed that the symbionts have undergone genome rearrangements, and up to 35% of their genes may have been acquired by horizontal gene transfer. Many of the genes specific to the symbionts were homologs of virulence genes. We discovered an abundant and diverse array of genes similar to insecticidal toxins of nematode and aphid symbionts, and toxins of pathogens such as Yersinia and Vibrio. Transcriptomics and proteomics revealed that the SOX symbionts express the toxin-related genes (TRGs) in their hosts. We hypothesize that the symbionts use these TRGs in beneficial interactions with their host, including protection against parasites. This would explain why a mutualistic symbiont would contain such a remarkable 'arsenal' of TRGs.
Intestinal microbiota signatures associated with inflammation history in mice experiencing recurring colitis
2015 - Front Microbiol, 6: 1408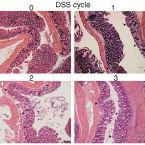
Abstract:
Acute colitis causes alterations in the intestinal microbiota, but the microbiota is thought to recover after such events. Extreme microbiota alterations are characteristic of human chronic inflammatory bowel diseases, although alterations reported in different studies are divergent and sometimes even contradictory. To better understand the impact of periodic disturbances on the intestinal microbiota and its compositional difference between acute and relapsing colitis, we investigated the beginnings of recurrent inflammation using the dextran sodium sulfate (DSS) mouse model of chemically induced colitis. Using bacterial 16S rRNA gene-targeted pyrosequencing as well as quantitative fluorescence in situ hybridization, we profiled the intestinal and stool microbiota of mice over the course of three rounds of DSS-induced colitis and recovery. We found that characteristic inflammation-associated microbiota could be detected in recovery-phase mice. Successive inflammation episodes further drove the microbiota into an increasingly altered composition post-inflammation, and signatures of colitis history were detectable in the microbiota more sensitively than by pathology analysis. Bacterial indicators of murine colitis history were identified in intestinal and stool samples, with a high degree of consistency between both sample types. Stool may therefore be a promising non-invasive source of bacterial biomarkers that are highly sensitive to inflammation state and history.
Following the footsteps of chlamydial gene regulation
2015 - Mol. Biol. Evol., 32: 3035-3046
Abstract:
Regulation of gene expression ensures an organism responds to stimuli and undergoes proper development. Although the regulatory networks in bacteria have been investigated in model microorganisms, nearly nothing is known about the evolution and plasticity of these networks in obligate, intracellular bacteria. The phylum Chlamydiae contains a vast array of host-associated microbes, including several human pathogens. The Chlamydiae are unique among obligate, intracellular bacteria as they undergo a complex biphasic developmental cycle in which large swaths of genes are temporally regulated. Coupled with the low number of transcription factors, these organisms offer a model to study the evolution of regulatory networks in intracellular organisms. We provide the first comprehensive analysis exploring the diversity and evolution of regulatory networks across the phylum. We utilized a comparative genomics approach to construct predicted coregulatory networks, which unveiled genus- and family-specific regulatory motifs and architectures, most notably those of virulence-associated genes. Surprisingly, our analysis suggests that few regulatory components are conserved across the phylum, and those that are conserved are involved in the exploitation of the intracellular niche. Our study thus lends insight into a component of chlamydial evolution that has otherwise remained largely unexplored.
Marine amoebae with cytoplasmic and perinuclear symbionts deeply branching in the Gammaproteobacteria
2015 - Sci Rep, 5: 13381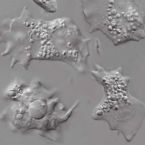
Abstract:
Amoebae play an important ecological role as predators in microbial communities. They also serve as niche for bacterial replication, harbor endosymbiotic bacteria and have contributed to the evolution of major human pathogens. Despite their high diversity, marine amoebae and their association with bacteria are poorly understood. Here we describe the isolation and characterization of two novel marine amoebae together with their bacterial endosymbionts, tentatively named 'Candidatus Occultobacter vannellae' and 'Candidatus Nucleophilum amoebae'. While one amoeba strain is related to Vannella, a genus common in marine habitats, the other represents a novel lineage in the Amoebozoa. The endosymbionts showed only low similarity to known bacteria (85-88% 16S rRNA sequence similarity) but together with other uncultured marine bacteria form a sister clade to the Coxiellaceae. Using fluorescence in situ hybridization and transmission electron microscopy, identity and intracellular location of both symbionts were confirmed; one was replicating in host-derived vacuoles, whereas the other was located in the perinuclear space of its amoeba host. This study sheds for the first time light on a so far neglected group of protists and their bacterial symbionts. The newly isolated strains represent easily maintainable model systems and pave the way for further studies on marine associations between amoebae and bacterial symbionts.
Prediction of microbial phenotypes based on comparative genomics
2015 - BMC Bioinformatics, 16: S1
Abstract:
The accessibility of almost complete genome sequences of uncultivable microbial species from metagenomes necessitates computational methods predicting microbial phenotypes solely based on genomic data. Here we investigate how comparative genomics can be utilized for the prediction of microbial phenotypes. The PICA framework facilitates application and comparison of different machine learning techniques for phenotypic trait prediction. We have improved and extended PICA's support vector machine plug-in and suggest its applicability to large-scale genome databases and incomplete genome sequences
Endosymbionts escape dead hydrothermal vent tubeworms to enrich the free-living population.
2015 - Proc. Natl. Acad. Sci. U.S.A., 36: 11300-11305
Abstract:
Theory predicts that horizontal acquisition of symbionts by plants and animals must be coupled to release and limited dispersal of symbionts for intergenerational persistence of mutualisms. For deep-sea hydrothermal vent tubeworms (Vestimentifera, Siboglinidae), it has been demonstrated that a few symbiotic bacteria infect aposymbiotic host larvae and grow in a newly formed organ, the trophosome. However, whether viable symbionts can be released to augment environmental populations has been doubtful, because (i) the adult worms lack obvious openings and (ii) the vast majority of symbionts has been regarded as terminally differentiated. Here we show experimentally that symbionts rapidly escape their hosts upon death and recruit to surfaces where they proliferate. Estimating symbiont release from our experiments taken together with well-known tubeworm density ranges, we suggest a few million to 1.5 billion symbionts seeding the environment upon death of a tubeworm clump. In situ observations show that such clumps have rapid turnover, suggesting that release of large numbers of symbionts may ensure effective dispersal to new sites followed by active larval colonization. Moreover, release of symbionts might enable adaptations that evolve within host individuals to spread within host populations and possibly to new environments.
Draft genome sequence of 'Candidatus Hepatoplasma crinochetorum Ps' – a bacterial symbiont in the hepatopancreas of the terrestrial isopod Porcellio scaber
2015 - Genome Announc, 3: e00674-15
Abstract:
'Candidatus Hepatoplasma crinochetorum Ps' is an extracellular symbiont residing in the hepatopancreas of the terrestrial isopod Porcellio scaber. Its genome is highly similar to that of the close relative 'Cand. Hepatoplasma crinochetorum Av' from Armadillidium vulgare. However, instead of a CRISPR/Cas system it encodes a type I restriction modification system.
Plastid establishment did not require a chlamydial partner
2015 - Nat Commun, 6: 6421
Abstract:
Primary plastids descend from the cyanobacterial endosymbiont of an ancient eukaryotic host, but the initial selective drivers that stabilized the association between these two cells are still unclear. One hypothesis that has achieved recent prominence suggests that the first role of the cyanobiont was in energy provision for a host cell whose reserves were being depleted by an intracellular chlamydial pathogen. A pivotal claim is that it was chlamydial proteins themselves that converted otherwise unusable cyanobacterial metabolites into host energy stores. We test this hypothesis by investigating the origins of the key enzymes using sophisticated phylogenetics. Here we show a mosaic origin for the relevant pathway combining genes with host, cyanobacterial or bacterial ancestry, but we detect no strong case for Chlamydiae to host transfer under the best-fitting models. Our conclusion is that there is no compelling evidence from gene trees that Chlamydiae played any role in establishing the primary plastidendosymbiosis.
Emendation of the family Chlamydiaceae: Proposal of a single genus, Chlamydia, to include all currently recognized species
2015 - Syst Appl Microbiol, 38: 99-103
Abstract:
The family Chlamydiaceae (order Chlamydiales, phylum Chlamydiae) comprises important, obligate intracellular bacterial pathogens of humans and animals. Subdivision of the family into the two genera Chlamydia and Chlamydophila has been discussed controversially during the past decade. Here, we have revisited the current classification in the light of recent genomic data and in the context of the unique biological properties of these microorganisms. We conclude that neither generally used 16S rRNA sequence identity cut-off values nor parameters based on genomic similarity consistently separate the two genera. Notably, no easily recognizable phenotype such as host preference or tissue tropism is available that would support a subdivision. In addition, the genus Chlamydophila is currently not well accepted and not used by a majority of research groups in the field. Therefore, we propose the classification of all 11 currently recognized Chlamydiaceae species in a single genus, the genus Chlamydia. Finally, we provide emended descriptions of the family Chlamydiaceae, the genus Chlamydia, as well as the species Chlamydia abortus, Chlamydia caviae and Chlamydia felis.
A nanoscale secondary ion mass spectrometry study of dinoflagellate functional diversity in reef-building corals.
2015 - Environ. Microbiol., 10: 3570-80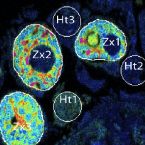
Abstract:
Nutritional interactions between corals and symbiotic dinoflagellate algae lie at the heart of the structural foundation of coral reefs. Whilst the genetic diversity of Symbiodinium has attracted particular interest because of its contribution to the sensitivity of corals to environmental changes and bleaching (i.e. disruption of coral-dinoflagellate symbiosis), very little is known about the in hospite metabolic capabilities of different Symbiodinium types. Using a combination of stable isotopic labelling and nanoscale secondary ion mass spectrometry (NanoSIMS), we investigated the ability of the intact symbiosis between the reef-building coral Isopora palifera, and Symbiodinium C or D types, to assimilate dissolved inorganic carbon (via photosynthesis) and nitrogen (as ammonium). Our results indicate that Symbiodinium types from two clades naturally associated with I. palifera possess different metabolic capabilities. The Symbiodinium C type fixed and passed significantly more carbon and nitrogen to its coral host than the D type. This study provides further insights into the metabolic plasticity among different Symbiodinium types in hospite and strengthens the evidence that the more temperature-tolerant Symbiodinium D type may be less metabolically beneficial for its coral host under non-stressful conditions.
Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at